|
Как и когда зарождается болезнь Хантингтона
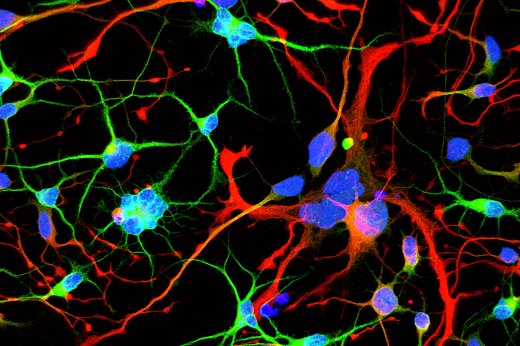
Нейроны, развивающиеся из человеческих эмбриональных стволовых клеток с отредактированным геном белка хантингтина, обнаруживают признаки серьезных нарушений, таких как множественные ядра (синие), задолго до появления симптомов заболевания. (Фото: Laboratory of Stem Cell Biology and Molecular Embryology at The Rockefeller University) (Увеличить)
«Мы должны переосмыслить подход к лечению болезни Хантингтона»
Симптомы болезни Хантингтона обычно проявляются в среднем возрасте, но новое исследование показывает, что нейронные аномалии обнаруживаются уже на первых этапах эмбрионального развития. Полученные недавно данные свидетельствуют о том, что лечение этого заболевания необходимо начинать как можно раньше.
Проведенные недавно исследования, возможно, способны вывести ученых из тупика в изучении болезни Хантингтона и принести успех в поисках мишеней для лечения этого наследственного нейродегенеративного заболевания.
У большинства пациентов с болезнью Хантингтона симптомы начинают проявляться в среднем возрасте. К таковым относятся судорожные движения и, как следствие все более интенсивной потери нейронов головного мозга, слабоумие. Но данные, полученные в ходе одного из новых исследований, позволяют предположить, что эти симптомы могут быть поздним проявлением заболевания, возникающего гораздо раньше, на первых этапах эмбрионального развития.
Группа ученых из Рокфеллеровского университета (The Rockefeller University) во главе с профессором Али Бриванлу (Ali Brivanlou), PhD, разработала первую систему для моделирования болезни Хантингтона, основанную на человеческих эмбриональных стволовых клетках. В статье, опубликованной в журнале Development, они описали ранние аномалии нейронов и то, как эти клетки образуют большие структуры, ранее не связываемые с этим заболеванием.
По мнению доктора Бриванлу, результаты его исследования подтверждают идею о том, то первый толчок к развитию болезнь Хантингтона получает сразу после оплодотворения. Дальше изменения идут по цепочке. Наибольшего проявления последствия этих изменений достигают через десятилетия, когда симптомы заболевания манифестируются со всей очевидностью.
Результаты исследования группы профессора Бриванлу могут помочь найти наилучший подход к лечению этого нейродегенеративного заболевания и в конечном итоге привести к появлению эффективных лекарственных препаратов.
Болезнь Хантингтона – одно из немногих заболеваний, о которых точно известно, что их виновником является генетика. Болезнь развивается у ста процентов людей с мутантной формой гена, кодирующего белок хантингтин (Huntingtin, HTT). Мутация заставляет этот ген продуцировать более длинный, чем в норме, белок. Сама ДНК представляет собой повторяющуюся последовательность из трех нуклеотидов (CAG), и чем больше таких повторов, тем раньше начинается заболевание. [Количество кодонов CAG в дикой форме гена может быть разным, и существует некий его предел, ниже которого заболевание не развивается.]
До сих пор болезнь Хантингтона в основном изучалась на животных моделях заболевания, и многие ключевые вопросы оставались без ответа. В частности, ученые пока так и не смогли выяснить, какова функция белка хантингтина в норме и как именно его мутация создает проблемы в мозге.
Подозревая, что у человека, чей мозг намного больше и сложнее, чем у лабораторных животных, болезнь развивается по-другому, профессор Бриванлу и его коллеги разработали для своих исследований систему на человеческих клетках. Для получения серии линий человеческих эмбриональных стволовых клеток, отличавшихся только количеством повторов ДНК на концах HTT-генов, они использовали технологию редактирования генов CRISPR.
«То, что мы увидели, было полной неожиданностью», - делится впечатлениями доктор Бриванлу. «В клеточных линиях с мутантным HTT мы увидели гигантские клетки. Это выглядело как джунгли дезорганизации».
При делении клеток в них, как правило, сохраняется одно ядро. Однако в некоторых из этих более крупных мутировавших клеток содержалось до 12 ядер, что указывало на нарушение нейрогенеза, то есть генерации новых нейронов. Нарушение было прямо пропорционально количеству повторов, присутствовавших в мутации: чем больше повторов, тем больше многоядерных нейронов.
«Наша работа еще раз доказывает, что в этой патологии существует нераспознанный аспект развития», - считает профессор Бриванлу. «Возможно, болезнь Хантингтона, не только нейродегенеративное, но и нейроонтогенетическое заболевание».

Профессор Али Бриванлу (Ali Brivanlou), PhD. (Фото: Rockefeller University) (Увеличить)
Лечение болезни Хантингтона обычно ограничивается блокированием активности мутантного белка хантингтина. Ученые при этом основываются на предположении, что измененная форма белка НТТ более активна и поэтому токсична для нейронов. Однако работа Бриванлу показывает, что нарушение мозга может быть связано с отсутствием активности хантингтина.
Чтобы прояснить его функцию, исследователи создали клеточные линии с полным отсутствием хантингтина. Эти клетки оказались очень похожими на те, что связаны с патологией Хантингтона, что подтверждает мысль о том, что к болезни ведет не избыток данного белка, а его недостаток.
Это важные результаты, отмечает проф. Бриванлу, так как они показывают, что существующие методы лечения, направленные на блокирование активности хантингтина, на самом деле могут принести больше вреда, чем пользы.
«Мы должны переосмыслить подход к лечению болезни Хантингтона, - считает ученый. «И роль белка HTT, и сроки начала лечения должны быть пересмотрены. К тому времени, когда у пациента проявляются симптомы, лечить может оказаться слишком поздно. Нам нужно вернуться к самым ранним событиям, которые вызывают цепную реакцию, приводящую в конечном итоге к болезни, и сделать мишенью новых препаратов причину, а не следствие».
Исследователи надеются, что их новые клеточные линии станут полезным ресурсом для изучения клеточных и молекулярных тонкостей болезни Хантингтона и полагают, что они могут служить моделью для изучения и других заболеваний головного мозга, свойственных только человеку.
По материалам
Uncovering the early origins of Huntington's disease
Оригинальная статья:
Albert Ruzo, Gist F. Croft, Jakob J. Metzger, Szilvia Galgoczi, Lauren J. Gerber, Cecilia Pellegrini, Hanbin Wang, Jr, Maria Fenner, Stephanie Tse, Adam Marks, Corbyn Nchako, Ali H. Brivanlou. Chromosomal instability during neurogenesis in Huntington's disease
Related Articles: |